Surface charge
Surface charge is the electric charge present at an interface. There are many different processes which can lead to a surface being charged, including adsorption of ions, protonation/deprotonation, and the application of an external electric field. Surface charge causes a particle to emit an electric field, which causes particle repulsions and attractions, and is responsible for many colloidal properties.[1]
Contents |
Surface charge density
Surface charge density is defined as the amount of electric charge, q, that is present on a surface of given area, A[2]:
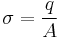
Conductors
According to Gauss’s law, a conductor at equilibrium carrying an applied current has no charge on its interior. Instead, the entirety of the charge of the conductor resides on the surface, and can be expressed by the equation:
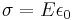 Where E is the electric field caused by the current in the conductor and
Where E is the electric field caused by the current in the conductor and 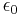 is the permittivity of the free space. It’s worth noting that this equation is only strictly accurate for conductors with infinitely large area, but it provides a good approximation if E is measured at the surface of the conductor.[3]
is the permittivity of the free space. It’s worth noting that this equation is only strictly accurate for conductors with infinitely large area, but it provides a good approximation if E is measured at the surface of the conductor.[3]
Colloids and immersed objects
| Compound | Chemical Formula | Point of Zero Charge |
|---|---|---|
| tungsten(VI) oxide | WO3 | 0.2-0.5[4] |
| silicon carbide (alpha) | SiC | 2-3.5[5] |
| manganese(IV) oxide | MnO2 | 4-5[4] |
| silicon nitride | Si3N4 | 6-7[6] |
| thallium(I) oxide | Tl2O | 8[7] |
| copper(II) oxide | CuO | 9.5[5] |
| nickel(II) oxide | NiO | 10-11[5] |
When a surface is immersed in a solution containing electrolytes, it develops a net surface charge. This is often because of ionic adsorption. Aqueous solutions universally contain positive and negative ions (cations and anions, respectively), which interact with partial charges on the surface, adsorbing to and thus ionizing the surface and creating a net surface charge.[8] This net charge results in a surface potential [L], which causes the surface to be surrounded by a cloud of counter-ions, which extends from the surface into the solution, and also generally results in repulsion between particles. The larger the partial charges in the material, the more ions are adsorbed to the surface, and the larger the cloud of counter-ions. A solution with a higher concentration of electolytes also increases the size of the counter-ion cloud. This ion/counterion layer is known as the electric double layer. [9]
A solution's pH can also greatly effect surface charge because functional groups present on the surface of particles can often contain oxygen or nitrogen, two atoms which can be protonated or deprotonated to become charged. Thus, as the concentration of hydrogen ions changes, so does the surface charge of the particles. At a given pH, the average surface charge will be equal to zero; this is known as the point of zero charge (PCZ).[1] A list of common substances and their associated PCZs is shown to the right.
Interfacial potential
An interface is defined as the common boundary formed between two different phases, such as between a solid and gas.[1] Electric potential, or charge, is the result of an object's capacity to be moved in an electric field. An interfacial potential is thus defined as a charge located at the common boundary between two phases (for example, an amino acid such as glutamate on the surface of a protein can have its side chain carboxylic acid deprotonated in environments with pH greater than 4.1 to produce a charged amino acid at the surface, which would create an interfacial potential). Interfacial potential is responsible for the formation of the electric double layer, which has a broad range of applications in what is termed electrokinetic phenomena. The development of the theory of the electric double layer is described below.
Helmholtz
The model dubbed the 'electric double layer' was first introduced by Hermann von Helmholtz. It assumes that a solution is only composed of electrolytes, no reactions occur near the electrode which could transfer electrons, and that the only Van der Waals interactions are present between the ions in solution and the electrode. These interactions arise only due to the charge density associated with the electrode which arises from either an excess or deficiency of electrons at the electrode's surface. To maintain electrical neutrality the charge of the electrode will be balanced by a redistribution of ions close to it's surface. The attracted ions thus form a layer balancing the electrode's charge. The closest distance an ion can come to the electrode will be limited to the radius of the ion plus a single solvation sphere around an individual ion. Overall, two layers of charge and a potential drop from the electrode to the edge of the outer layer (outer Helmholtz Plane) are observed. Given the above description, the Helmholtz model is equivalent in nature to an electrical capacitor with two separated plates of charge, for which a linear potential drop is observed at increasing distance from the plates.
The Helmholtz model, while a good foundation for the description of the interface does not take into account several important factors: diffusion/mixing in solution, the possibility of adsorption on to the surface and the interaction between solvent dipole moments and the electrode. [10]
Gouy-Chapman
Gouy-Chapman theory describes the effect of a static surface charge on a surface's potential.[11] "Gouy suggested that interfacial potential at the charged surface could be attributed to the presence of a number of ions of given charge attached to its surface, and to an equal number of ions of opposite charge in the solution."[12] A positive surface charge will form a double layer, since negative ions in solution tend to balance the positive surface charge. Counter ions are not rigidly held, but tend to diffuse into the liquid phase until the counter potential set up by their departure restricts this tendency. The kinetic energy of the counter ions will, in part, affect the thickness of the resulting diffuse double layer. The relation between C, the counter ion concentration at the surface, and 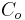 , the counter ion concentration in the external solution, is the Boltzmann factor:
, the counter ion concentration in the external solution, is the Boltzmann factor:
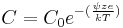 Where z is the charge on the ion, e is the charge of a proton, k is the Boltzmann constant and ψ is the potential of the charged surface. This however is inaccurate close to the surface, because it assumes that molar concentration is equal to activity. It also assumes that ions were modeled as point charges and was later modified. An improvement of this theory, known as the modified Gouy-Chapman theory, included the finite size of the ions with respect to their interaction with the surface in the form of a plane of closest approach.[13]
Where z is the charge on the ion, e is the charge of a proton, k is the Boltzmann constant and ψ is the potential of the charged surface. This however is inaccurate close to the surface, because it assumes that molar concentration is equal to activity. It also assumes that ions were modeled as point charges and was later modified. An improvement of this theory, known as the modified Gouy-Chapman theory, included the finite size of the ions with respect to their interaction with the surface in the form of a plane of closest approach.[13]
Surface charge & surface potential
The relation between surface charge and surface potential can be expressed by the Grahame equation, derived from the Gouy-Chapman theory by assuming the electroneutrality condition, which states that the total charge of the double layer must be equal to the negative of the surface charge. Using the one dimensional Poisson equation and assuming that, at an infinitely great distance, the potential gradient is equal to 0, the Grahame equation is obtained[1] :
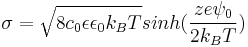
For the case of lower potentials, 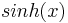 can be expanded to =
can be expanded to = 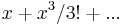
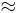
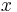 , and
, and 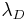 is defined as the Debye length. Which leads to the simple expression:
is defined as the Debye length. Which leads to the simple expression:
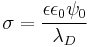
Stern
The Stern model of the double layer is essentially a combination of Helmholtz and Gouy-Chapman theories. His theory states that ions do have finite size, so cannot approach the surface closer than a few nanometers. Through a distance known as the Stern Layer, ions can be adsorbed onto the surface up to a point referred to as the slipping plane, where the ions adsorbed meet the bulk liquid. At the slipping plane the potential, Ψ, has decreased to what is known as the zeta potential. Although zeta potential is an intermediate value, it is sometimes considered to be more significant than surface potential as far as electrostatic repulsion is concerned.[1]
Applications of surface charge
Charged surfaces are extremely important and are used in many applications. For example, solutions of large colloidal particles depend almost entirely on repulsion due to surface charge in order to stay dispersed.[14] If these repulsive forces were to be disrupted, perhaps by the addition of a salt or a polymer, the colloidal particles would no longer be able to sustain suspension and would subsequently flocculate.[15]
Electrokinetic phenomena
Electrokinetic phenomena refers to a variety of effects resulting from an electrical double layer. A noteworthy example is electrophoresis, where a charged particle suspended in a media will move as a result of an applied electrical field.[16] Electrophoresis is widely used in biochemistry to distinguish molecules, such as proteins, based on size and charge. Other examples include electro-osmosis, sedimentation potential, and streaming potential.[1]
Proteins
Proteins often have groups present on their surfaces that can be ionized or deionized depending on pH, making it relatively easy to change the surface charge of a protein. This has particularly important ramifications on the activity of proteins that function as enzymes or membrane channels, mainly, that the protein's active site must have the right surface charge in order to be able to bind a specific substrate.[17]
Adhesives/coatings
Charged surfaces are often useful in creating surfaces that will not adsorb certain molecules (for example, in order to prevent the adsorption of basic proteins, a positively charged surface should be used). Polymers are very useful in this respect in that they can be functionalized so that they contain ionizable groups, which serve to provide a surface charge when submerged in an aqueous solution.[18]
References
- ^ a b c d e f g Hans-Jurgen, Butt; Graf, Karlheinz; Kappl, Michael (2006). Physics and chemistry of interfaces. Germany: Wiley-VCH. pp. 45, 55, 56, 76–82. ISBN 978-3-527-40629-6.
- ^ Weisstein, Eric W. (2007). "Surface charge density". Wolfram Research. http://scienceworld.wolfram.com/physics/SurfaceChargeDensity.html. Retrieved 30 May 2011.
- ^ Nave, Carl R. (2010). "Gaussian surfaces". Georgia State University. http://hyperphysics.phy-astr.gsu.edu/hbase/electric/gausur.html. Retrieved 27 April 2011.
- ^ a b Marek Kosmulski, "Chemical properties of material surfaces", Marcel Dekker, 2001. Retrieved 30 May 2011
- ^ a b c Lewis, JA (2000). 'Colloidal processing of ceramics', Journal of the American Ceramic Society vol. 83, no. 10, pp.2341-2359. Retrieved 30 May 2011
- ^ Jolivet J.P., Metal oxide chemistry and synthesis. From solution to solid state, John Wiley & Sons Ltd. 2000,ISBN 0-471-97056-5 (English translation of the original French text, De la solution à l'oxyde, InterEditions et CNRS Editions, Paris, 1994). Retrieved 30 May 2011
- ^ Kosmulski M and Saneluta C (2004). 'Point of zero charge/isoelectric point of exotic oxides: Tl2O3', Journal of Colloid and Interface Science vol. 280, no. 2, pp. 544-545. Retrieved 30 May 2011
- ^ "Origins of surface charge". Silver Colloids. 2010. http://www.silver-colloids.com/Tutorials/Intro/pcs13.html. Retrieved 27 April 2011.
- ^ "The electric double layer". Silver Colloids. 2010. http://www.silver-colloids.com/Tutorials/Intro/pcs17A.html. Retrieved 27 April 2011.
- ^ "The electrical double layer". 2011. http://www.cartage.org.lb/en/themes/sciences/Chemistry/Electrochemis/Electrochemical/ElectricalDouble/ElectricalDouble.htm. Retrieved 27 April 2011.
- ^ Ehrenstein, Gerald (200). "Surface charge". http://www.biophysics.org/Portals/1/PDFs/Education/ehrenstein.pdf. Retrieved 30 May 2011.
- ^ SMIRNOV, Gerald (2011). "Double bilayer". http://web.nmsu.edu/~snsm/classes/chem435/Lab14/double_layer.html. Retrieved 30 May 2011.
- ^ Greathouse, Jeffery A.; Feller, Scott E.; McQuarrie, Donald A. (February 25, 1994). "The modified Gouy-Chapman theory: Comparisons between electrical double layer models of clay swelling". http://???. Retrieved 30 May 2011.
- ^ "Zeta potential measurement". Brookhaven Instruments Ltd.. 2007. http://www.brookhaven.co.uk/zeta-potential.html. Retrieved 16 Apr. 2011.
- ^ Hubbe, Martin (2007). "Flocculation of colloids or of fiber slurries". North Carolina State University. http://www4.ncsu.edu/~hubbe/EqipUnit/Floccula.htm. Retrieved 16 Apr. 2011.
- ^ "Chapter 4: Electrophoresis - Introduction". Dr. William H. Heidcamp, Biology Department, Gustavus Adolphus College. 1995. http://homepages.gac.edu/~cellab/chpts/chpt4/intro4.html. Retrieved 30 May 2011.
- ^ Escobar, Laura; Root, Michael J.; MacKinnon, Robert (July 1993). "Influence of protein surface charge on the bimolecular kinetics of a potassium channel peptide inhibitor". Biochemistry 32 (27): 6982–6987. doi:10.1021/bi00078a024. PMID 7687466. http://pubs.acs.org/doi/pdf/10.1021/bi00078a024. Retrieved 16 April 2011. http://pubs.acs.org/doi/pdf/10.1021/bi00078a024
- ^ Haselberg, Rob; van der Sneppen, Lineke; Ariese, Freek; Ubachs, Wim; Gooijer, Cees; de Jong, Gerhardus J.; Somsen, Govert W. (18 Nov. 2009). "Effectiveness of charged non-covalent polymer coatings against protein adsorption to silica surfaces studied by evanescent-wave cavity ring-down spectroscopy and capillary electrophoresis". Analytical Chemistry 81 (24): 10172–10178. doi:10.1021/ac902128n. http://pubs.acs.org/doi/pdf/10.1021/ac902128n. Retrieved 16 April 2011.